What Is Used To Determine Phylogeny
Muz Play
Mar 26, 2025 · 7 min read
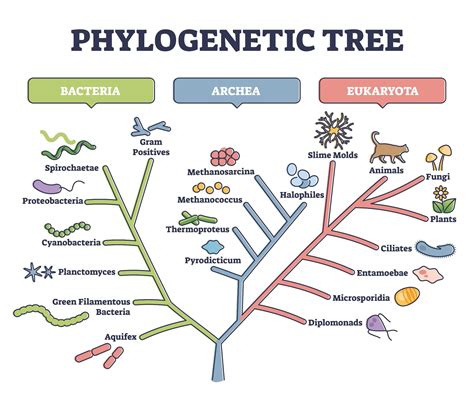
Table of Contents
What is Used to Determine Phylogeny? Unveiling the Evolutionary History of Life
Phylogeny, the evolutionary history and relationships among individuals or groups of organisms, is a cornerstone of modern biology. Understanding phylogenetic relationships allows us to reconstruct the tree of life, tracing the ancestry of species and revealing the patterns of diversification that have shaped the biodiversity we see today. But how do scientists actually determine phylogeny? It's a complex process that relies on a variety of data and sophisticated analytical techniques. This article delves into the core methods and data types used to build phylogenetic trees, highlighting the strengths and limitations of each approach.
The Foundation: Data Types for Phylogenetic Inference
The construction of a phylogenetic tree, also known as a cladogram or dendrogram, depends heavily on the type of data used. Different data sets provide different levels of resolution and can be more or less susceptible to errors. The most commonly used data types include:
1. Morphological Data: The Traditional Approach
For decades, morphological data – observable physical characteristics of organisms – were the primary source of information for building phylogenetic trees. This includes:
- Anatomical features: The presence, absence, or modification of structures like bones, organs, limbs, and other body parts. For example, the presence of wings in birds and bats, despite their very different evolutionary origins, can be used to infer relationships. However, convergent evolution (independent evolution of similar traits in unrelated lineages) can complicate this analysis.
- Embryological development: Similarities in developmental patterns can reveal shared ancestry. For example, the presence of pharyngeal arches in vertebrate embryos, regardless of the adult form, suggests a common ancestor.
- Fossil evidence: The fossil record provides crucial insights into extinct organisms and their relationships to extant (living) species. However, the fossil record is incomplete, and the interpretation of fossil morphology can be challenging.
Limitations of Morphological Data:
- Subjectivity: Interpreting morphological characteristics can be subjective, leading to discrepancies between researchers.
- Convergent evolution: Similar traits can evolve independently in different lineages due to similar environmental pressures. This can lead to inaccurate inferences of relatedness.
- Homoplasy: This refers to similar traits that do not share a common evolutionary origin. Homoplasy, including convergent evolution and evolutionary reversals (loss of a derived trait), can obscure true phylogenetic relationships.
- Limited resolution: Morphological data might not offer sufficient resolution to distinguish closely related species, especially in groups with subtle morphological differences.
2. Molecular Data: A Revolution in Phylogenetics
The advent of molecular biology revolutionized phylogenetics. Molecular data, including DNA, RNA, and protein sequences, provide a wealth of information for reconstructing evolutionary relationships:
- DNA sequences: These are the most widely used molecular data for phylogeny. Comparing DNA sequences from different organisms reveals the number of genetic differences between them, which can be used to infer evolutionary distance. Different regions of the genome evolve at different rates, allowing researchers to choose appropriate markers depending on the timescale being investigated.
- RNA sequences: Similar to DNA, RNA sequences can be used to infer phylogenetic relationships. Ribosomal RNA (rRNA) is particularly useful due to its slow rate of evolution, making it suitable for comparing distantly related organisms.
- Protein sequences: Proteins are the products of genes, and their amino acid sequences reflect the underlying DNA sequence. Protein sequences are useful for phylogenetic inference, particularly when analyzing highly conserved proteins.
Advantages of Molecular Data:
- Objectivity: Molecular data is more objective than morphological data, reducing subjectivity in phylogenetic inference.
- Large datasets: Molecular data allows for the analysis of large datasets, providing higher resolution and statistical power.
- Wider taxonomic coverage: Molecular data can be used to compare organisms that are morphologically very different or lack a detailed fossil record.
- Quantifiable differences: Molecular differences can be precisely quantified, allowing for more rigorous statistical analysis.
Limitations of Molecular Data:
- Homoplasy: Molecular data can also be subject to homoplasy, including convergent evolution and parallel evolution (independent evolution of similar traits with similar genetic changes).
- Horizontal gene transfer: In prokaryotes and some eukaryotes, genes can be transferred horizontally between unrelated organisms, complicating phylogenetic reconstruction. This complicates the use of single gene phylogenies and emphasizes the use of multi-gene approaches.
- Genome size: Analyzing complete genomes can be computationally demanding, even with modern computer technology.
- Data selection bias: The choice of genes or genomic regions can significantly influence the resulting phylogenetic tree.
Methods for Phylogenetic Inference
Once data is collected, various methods are used to construct phylogenetic trees. These methods differ in their approaches and assumptions:
1. Character-Based Methods: Parsimony and Maximum Likelihood
- Parsimony: This method seeks the simplest explanation for the observed data, choosing the tree that requires the fewest evolutionary changes to explain the observed character states. It's intuitively appealing but can be computationally intensive for large datasets.
- Maximum Likelihood: This method estimates the probability of observing the data given a particular tree and model of evolution. It considers the rates of character change and other evolutionary processes, making it more statistically robust than parsimony.
2. Distance-Based Methods: Neighbor-Joining and UPGMA
These methods calculate a distance matrix representing the pairwise differences between organisms, then use algorithms to construct a tree that best reflects those distances.
- Neighbor-Joining: A commonly used algorithm that is relatively fast and efficient, even for large datasets.
- UPGMA (Unweighted Pair Group Method with Arithmetic Mean): A simpler algorithm that assumes a constant rate of evolution.
3. Bayesian Methods: Markov Chain Monte Carlo (MCMC)
Bayesian methods use prior probabilities and data to calculate posterior probabilities of different trees. MCMC is a computational technique used to explore the space of possible trees and estimate the probabilities of different phylogenetic hypotheses. Bayesian methods are powerful but require substantial computational resources.
Choosing the Right Approach: Considerations and Challenges
The choice of data and method depends on several factors:
- Research question: The specific question being addressed will influence the choice of data and method. For example, investigating the relationships between closely related species might require highly variable molecular markers, while studying deep evolutionary relationships might necessitate slower evolving genes or morphological characters.
- Available data: The type and amount of available data will constrain the choice of method.
- Computational resources: Some methods, such as Bayesian analysis, are computationally demanding and require significant processing power.
- Assumptions of the method: Different methods make different assumptions about the evolutionary process, and it's crucial to choose a method whose assumptions are compatible with the data being analyzed.
Evaluating Phylogenetic Trees: Assessing Confidence and Robustness
Phylogenetic trees are hypotheses, not definitive statements about evolutionary history. It's crucial to assess the confidence and robustness of a phylogenetic tree:
- Bootstrap analysis: A statistical method used to assess the confidence in the branching patterns of a phylogenetic tree. Bootstrap values indicate the percentage of times a particular branch appears in a set of randomly resampled datasets. High bootstrap values (generally above 70%) indicate strong support for a particular branch.
- Bayesian posterior probabilities: Bayesian methods provide posterior probabilities for each branch, representing the probability that a given branch is correct given the data and the model. High posterior probabilities (generally above 0.95) indicate strong support for a branch.
- Sensitivity analysis: Examining how the tree changes when different data sets or methods are used helps to assess the robustness of the phylogenetic hypothesis.
The Future of Phylogenetics: Integrating Data and Refining Methods
Phylogenetics continues to evolve, incorporating new data types and more sophisticated analytical methods. Integrative approaches, combining different data types (e.g., morphological and molecular data), are becoming increasingly common. Advances in computational biology and genomics are making it possible to analyze ever-larger datasets, leading to more accurate and detailed phylogenetic reconstructions. The continuous development and refinement of phylogenetic methods ensures a deeper and more nuanced understanding of the evolutionary history of life on Earth. Furthermore, ongoing research continually improves our understanding of the complexities of evolution, leading to more sophisticated models incorporated into phylogenetic analyses. This continuous evolution of methods and data integration is key to building increasingly robust and informative trees of life. This allows us to better understand the evolutionary processes driving biodiversity and the interrelationships between all living things.
Latest Posts
Latest Posts
-
How To Calculate Velocity From Flow Rate
Mar 29, 2025
-
Write The Iupac Names Of The Given Carboxylic Acids
Mar 29, 2025
-
Multiplication Of A Polynomial By A Monomial
Mar 29, 2025
-
The Graph Of A Quadratic Function Is Called
Mar 29, 2025
-
Is Chemistry The Study Of Matter
Mar 29, 2025
Related Post
Thank you for visiting our website which covers about What Is Used To Determine Phylogeny . We hope the information provided has been useful to you. Feel free to contact us if you have any questions or need further assistance. See you next time and don't miss to bookmark.
